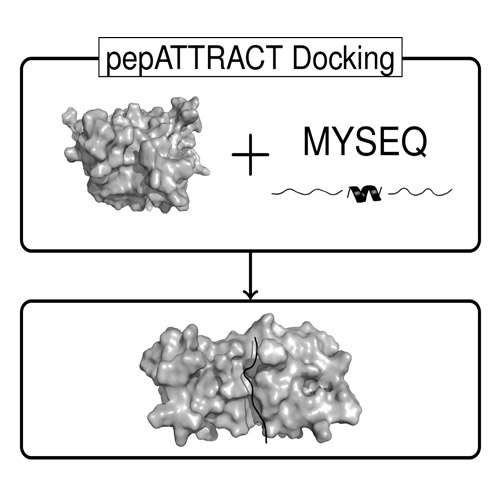

Fully Blind Peptide-Protein Docking with pepATTRACT
02-Jul-2015
Structure, Volume 23, Issue 8, p1507–1515, DOI: http://dx.doi.org/10.1016/j.str.2015.05.021
Peptide-protein interactions are ubiquitous in the cell and form an important part of the interactome. Computational docking methods can complement experimental characterization of these complexes, but current protocols are not applicable on the proteome scale. Here, we present a new fully blind flexible peptide-protein docking protocol, pepATTRACT, which combines a rapid coarse-grained global peptide docking search of the entire protein surface with a two-stage atomistic flexible refinement. Global unbound-unbound docking yielded near-native models for 70% of the docking cases when testing the protocol on the largest benchmark of peptide-protein complexes available to date. This performance is similar to that of state-of-the-art local docking protocols that rely on information about the binding site. Upon restricting the search to the peptide binding region, the resulting pepATTRACT-local approach outperformed existing methods.